







♦ What is Cystic Fibrosis_CF?
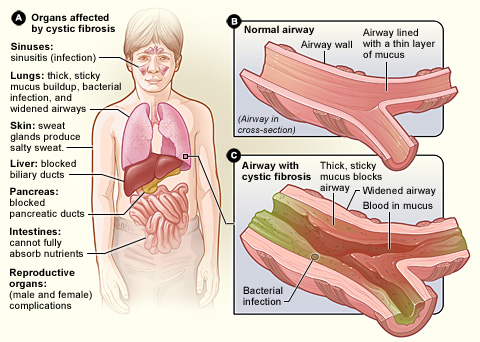
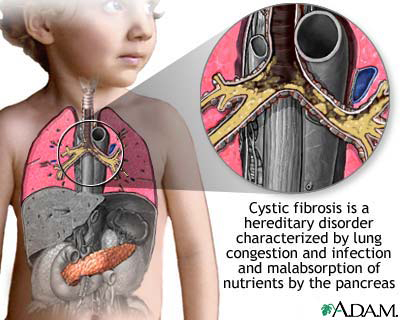
Is a genetic disorder that affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine.
♦ Signs and symptoms
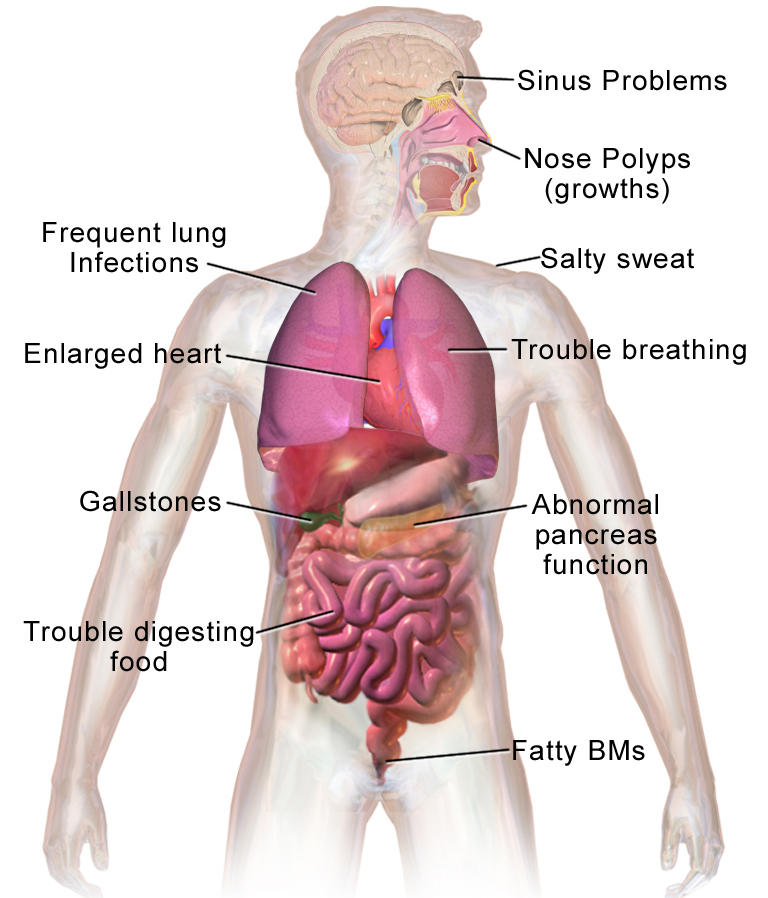
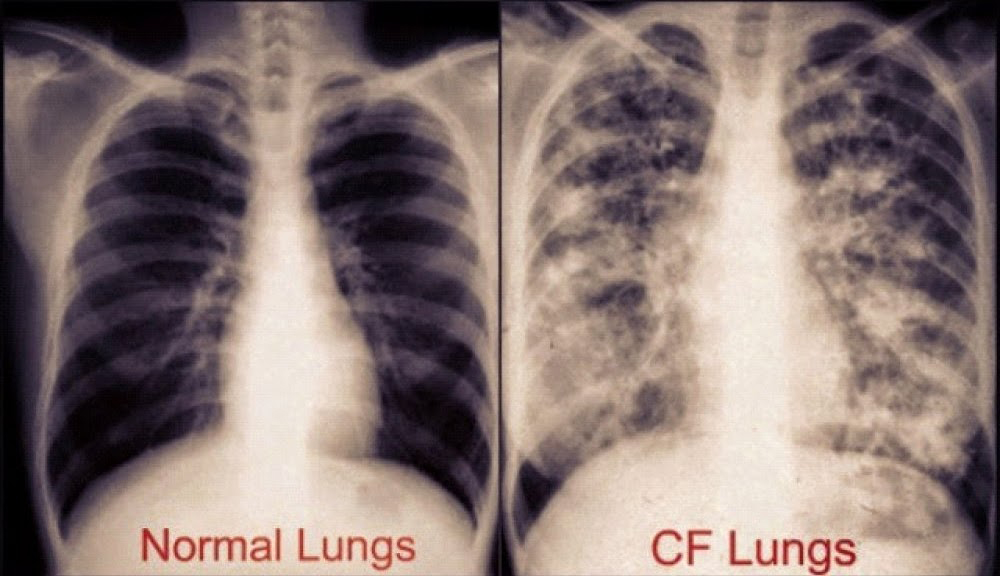
Long-term issues include difficulty breathing and coughing up mucus as a result of frequent lung infections.
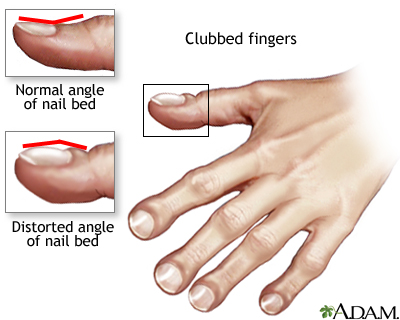
Other signs and symptoms may include sinus infections, poor growth, fatty stool, clubbing of the fingers and toes, and infertility in most males.
Different people may have different degrees of symptoms.
Clubbed fingers is a symptom of disease, often of the heart or lungs which cause chronically low blood levels of oxygen. Diseases which cause malabsorption, such as cystic fibrosis or celiac disease can also cause clubbing.
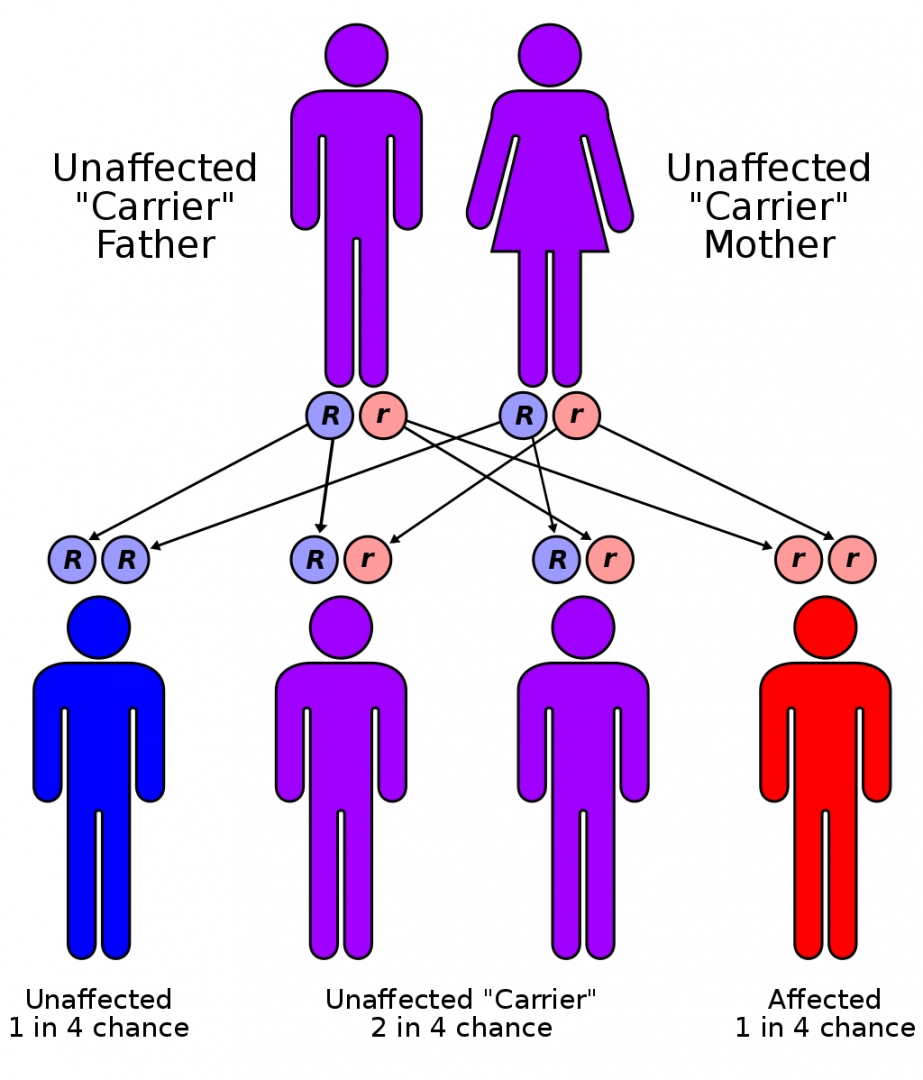
CF is inherited in an autosomal recessive manner. It is caused by the presence of mutations in both copies of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein.
Those with a single working copy are carriers and otherwise mostly normal.
CFTR is involved in production of sweat, digestive fluids, and mucus. When the CFTR is not functional, secretions which are usually thin instead become thick.
♦ Diagnosis
The condition is diagnosed by a sweat test and genetic testing. Screening of infants at birth takes place in some areas of the world.
♦ Treatment
There is no known cure for cystic fibrosis.
► Antibiotics
Many people with CF are on one or more antibiotics at all times, even when healthy, to prophylactically suppress infection.
Antibiotics are absolutely necessary whenever pneumonia is suspected or a noticeable decline in lung function is seen, and are usually chosen based on the results of a sputum analysis and the person's past response. This prolonged therapy often necessitates hospitalization and insertion of a more permanent IV such as a peripherally inserted central catheter or Port-a-Cath.
Inhaled therapy with antibiotics such as tobramycin, colistin, and aztreonam is often given for months at a time to improve lung function by impeding the growth of colonized bacteria. Inhaled antibiotic therapy helps lung function by fighting infection, but also has significant drawbacks such as development of antibiotic resistance, tinnitus, and changes in the voice. Inhaled levofloxacin may be used to treat Pseudomonas aeruginosa in people with cystic fibrosis who are infected. The early management of Pseudomonas aeruginosa infection is easier and better, using nebulised antibiotics with or without oral antibiotics may sustain its eradication up to 2 years.
All these factors related to the antibiotics use, the chronicity of the disease, and the emergence of resistant bacteria demand more exploration for different strategies such as antibiotic adjuvant therapy.
♦ Infertility
Infertility affects both men and women. At least 97% of men with cystic fibrosis are infertile, but not sterile and can have children with assisted reproductive techniques. The main cause of infertility in men with CF is congenital absence of the vas deferens (which normally connects the testes to the ejaculatory ducts of the penis), but potentially also by other mechanisms such as causing no sperm, abnormally shaped sperm, and few sperm with poor motility. Many men found to have congenital absence of the vas deferens during evaluation for infertility have a mild, previously undiagnosed form of CF.
Around 20% of women with CF have fertility difficulties due to thickened cervical mucus or malnutrition. In severe cases, malnutrition disrupts ovulation and causes a lack of menstruation
♦ Prognosis
The prognosis for cystic fibrosis has improved due to earlier diagnosis through screening and better treatment and access to health care.
In 1959, the median age of survival of children with CF was six months.
In 2010, survival is estimated to be 37 years for women and 40 for men.
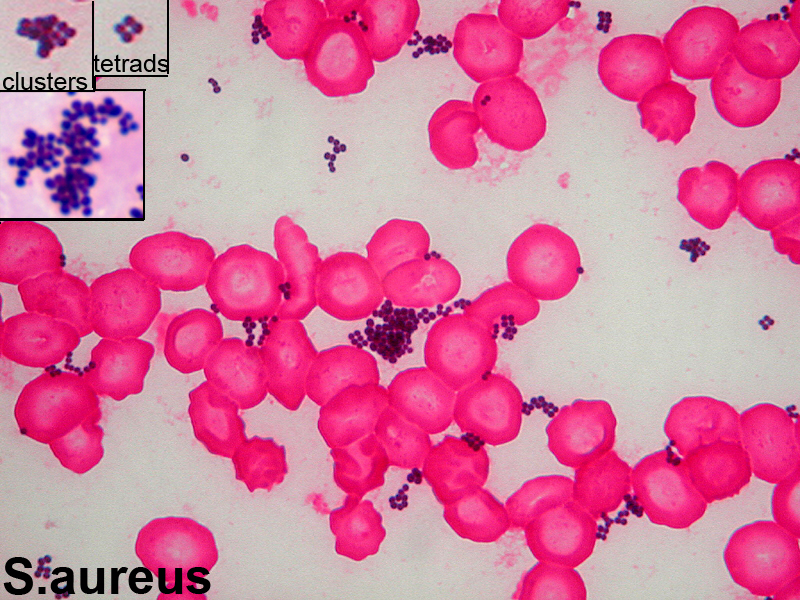
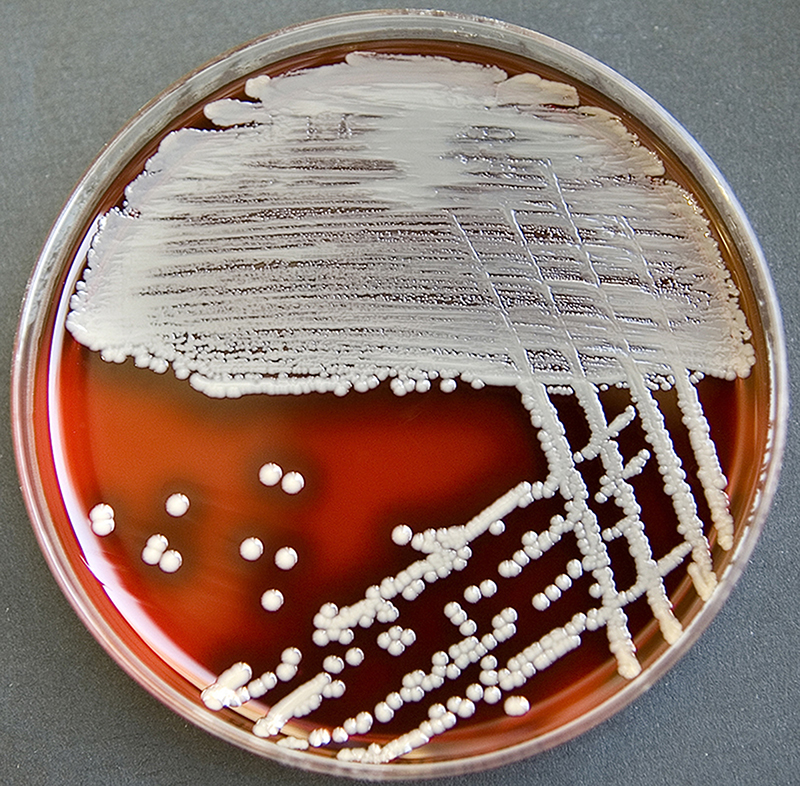
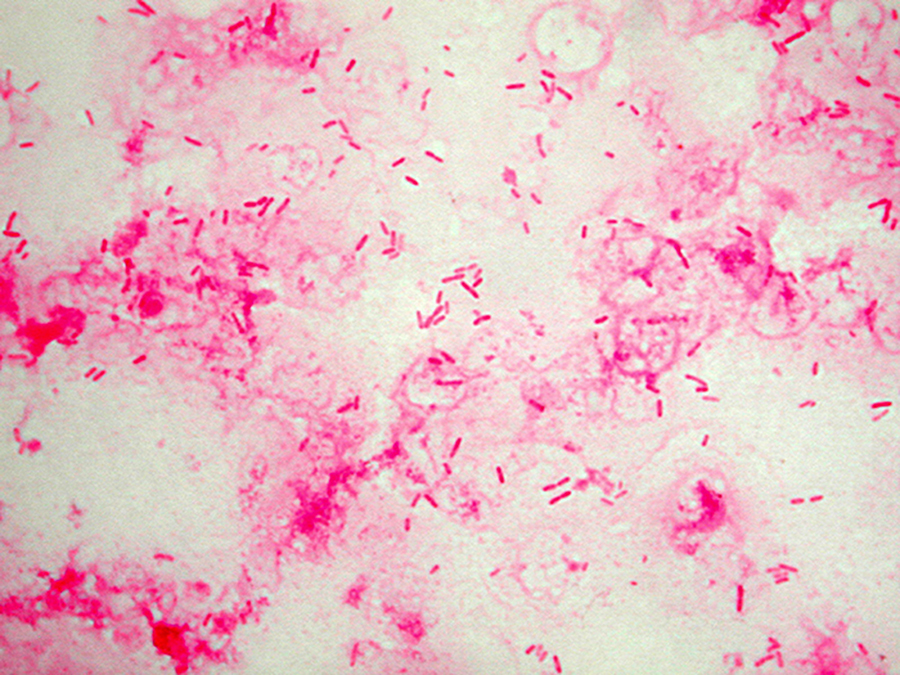
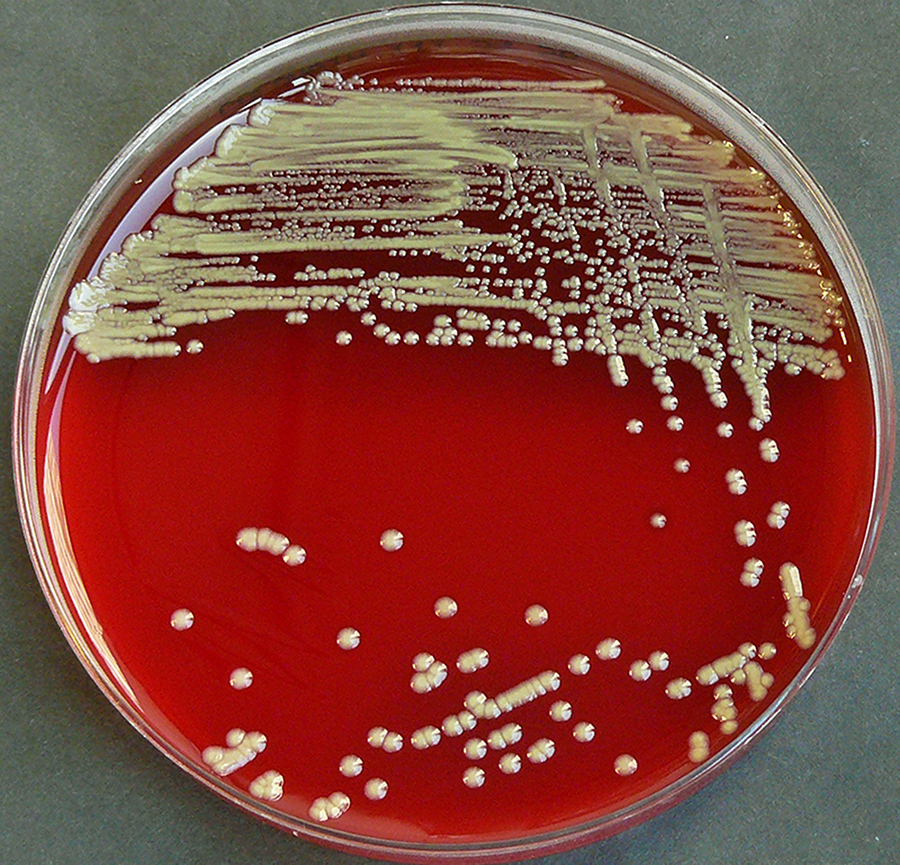
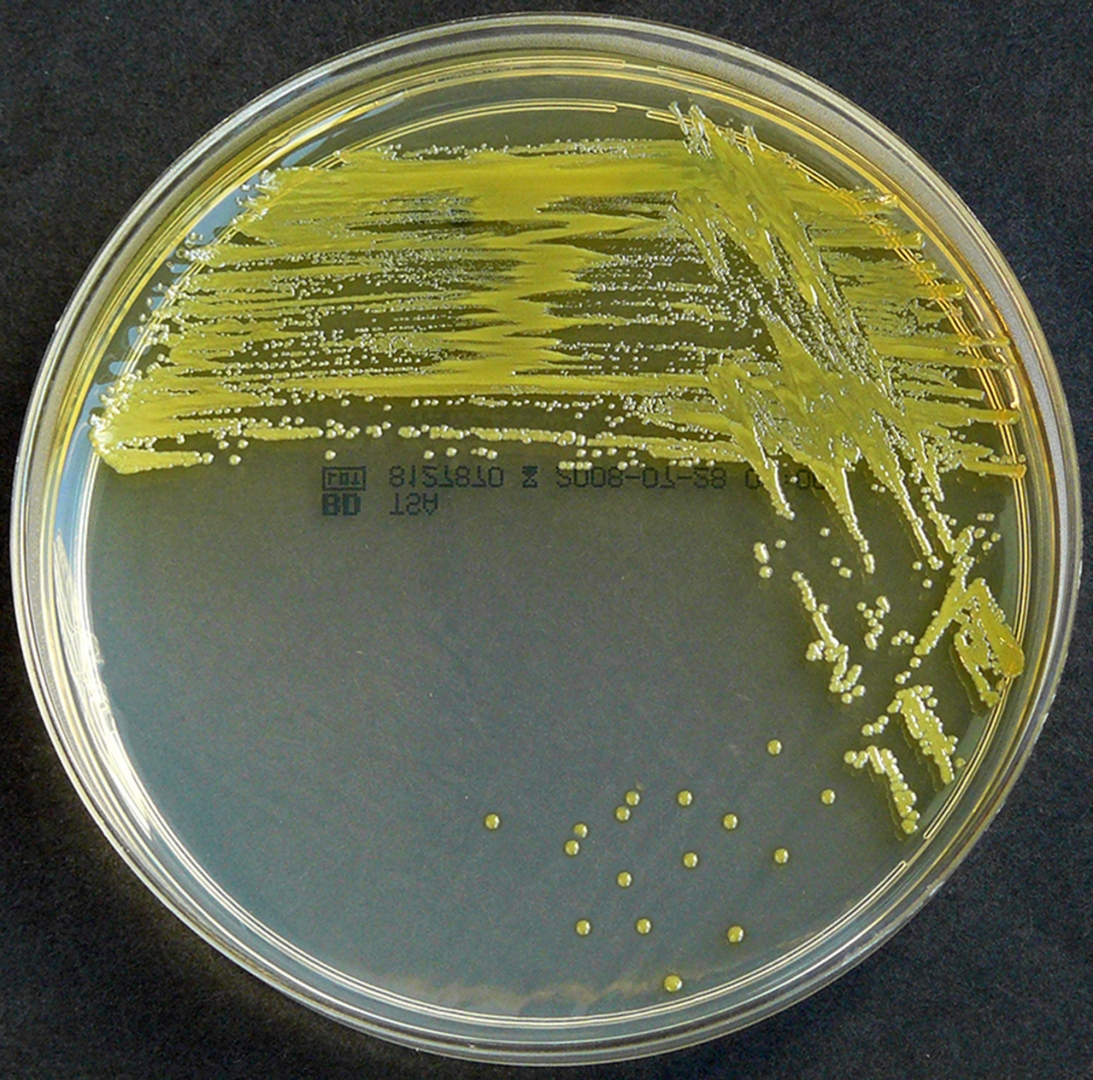
Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa are the three most common organisms causing lung infections in CF patients.
In addition to typical bacterial infections, people with CF more commonly develop other types of lung disease.
Among these is allergic bronchopulmonary aspergillosis, in which the body's response to the common fungus Aspergillus fumigatus causes worsening of breathing problems.
Another is infection with Mycobacterium avium complex, a group of bacteria related to tuberculosis, which can cause lung damage and does not respond to common antibiotics.
Over time, both the types of bacteria and their individual characteristics change in individuals with CF.
In the initial stage, common bacteria such as S. aureus and H. influenzae colonize and infect the lungs.
Eventually, Pseudomonas aeruginosa (and sometimes Burkholderia cepacia) dominates.
By 18 years of age, 80% of patients with classic CF harbor P. aeruginosa, and 3.5% harbor B. cepacia. Once within the lungs, these bacteria adapt to the environment and develop resistance to commonly used antibiotics.
Pseudomonas can develop special characteristics that allow the formation of large colonies, known as "mucoid" Pseudomonas, which are rarely seen in people who do not have CF.
Infection can spread by passing between different individuals with CF. In the past, people with CF often participated in summer "CF camps" and other recreational gatherings. Hospitals grouped patients with CF into common areas and routine equipment (such as nebulizers) was not sterilized between individual patients. This led to transmission of more dangerous strains of bacteria among groups of patients. As a result, individuals with CF are now routinely isolated from one another in the healthcare setting, and healthcare providers are encouraged to wear gowns and gloves when examining patients with CF to limit the spread of virulent bacterial strains.
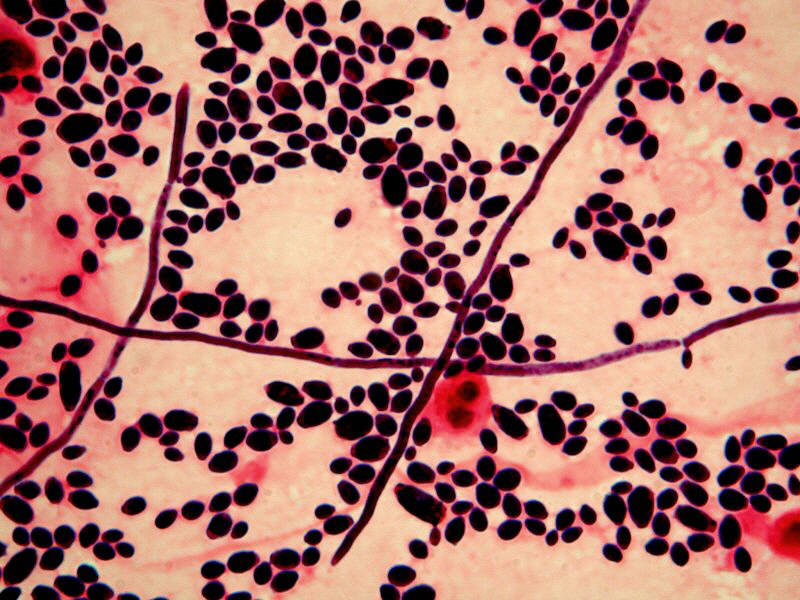
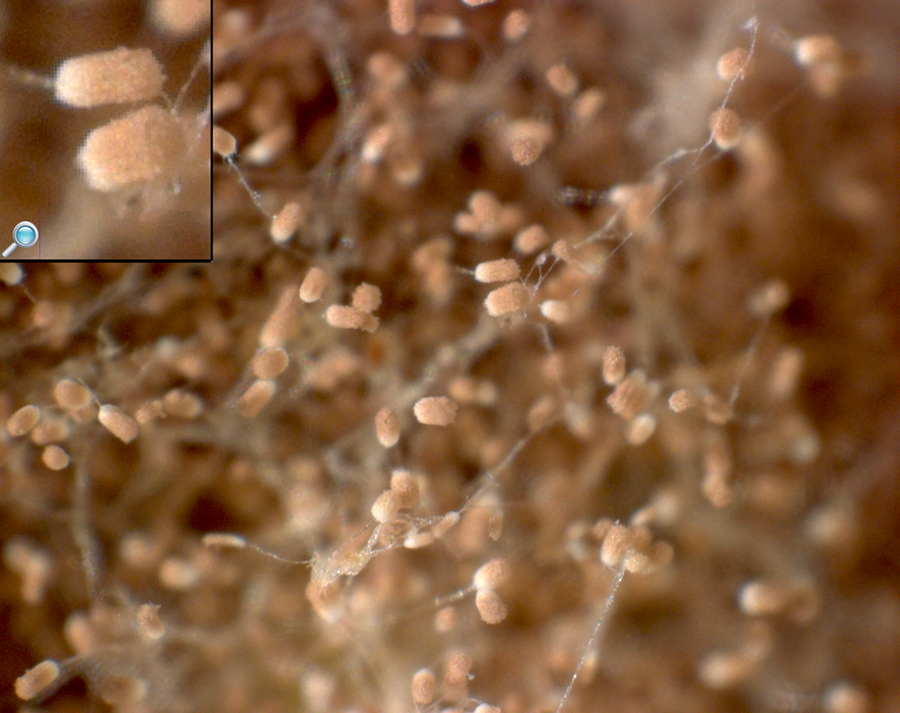
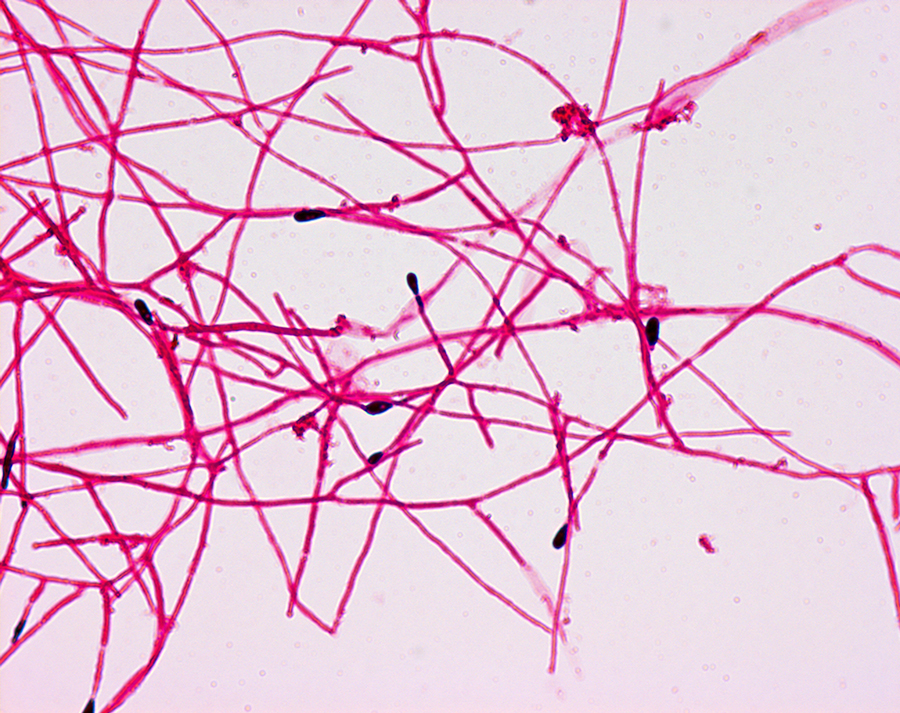
CF patients may also have their airways chronically colonized by filamentous fungi (such as Aspergillus fumigatus, Scedosporium apiospermum, Aspergillus terreus) and/or yeasts (such as Candida albicans); other filamentous fungi less commonly isolated include Aspergillus flavus and Aspergillus nidulans (occur transiently in CF respiratory secretions) and Exophiala dermatitidis and Scedosporium prolificans (chronic airway-colonizers); some filamentous fungi such as Penicillium emersonii and Acrophialophora fusispora are encountered in patients almost exclusively in the context of CF.

Cystic Fibrosis_CF
Related
References
wikipedia
Photos
wikipedia
MMIZ, ErasmusMC, Rotterdam_Loes van Damme
lungs: https://www.esiason.org/what-is-cf
http://www.mindsofmalady.com/2015/05/groundbreaking-treatment-for-patients.html
clubbing: https://medlineplus.gov/ency/imagepages/18127.htm
https://www.globalgiving.org/projects/cystic-fibrosis-is-not-a-death-sentence/
https://stock.adobe.com/search?k=cystic+fibrosis
https://stock.adobe.com/search?k=cystic+fibrosis&asset_id=229039543

- Actinomycosis
- Anthrax
- Biopsy Sinusitis_Aspergillus flavus
- Botulism
- Brucellosis
- Cat Scratch Disease
- Cellulitis
- Cholera
- Creutzfeldt-Jakob Disease
- Cystic Fibrosis_CF
- Diphtheria
- Erysipelas
- Erysipeloid or fish poison
- Legionnaires disease
- Lemierre syndrome
- Leprosy
- Listeriosis
- Lyme / Borreliosis
- Melioidosis
- Meningitis
- Plague
- Syphilis
- Tetanus
- Trench Mouth_Plaut-Vincent_acute necrotizing ulcerative gingivitis
- Tuberculosis (TB)
- Tularemia_Rabbit Fever
- Typhoid fever (Epidemic typhus)
- Whooping Cough